Self-assembled molecular layers strike a balance
07 Sep 2018 Anna Demming
Power is nothing without control, even for graphene. What attracted Ruben Perez, Professor of Condensed Matter Physics at Universidad Autonoma de Madrid, to study the self-assembly of molecular layers on graphene supported by various substances was the potential it opened up to tune graphene’s properties for technological applications.
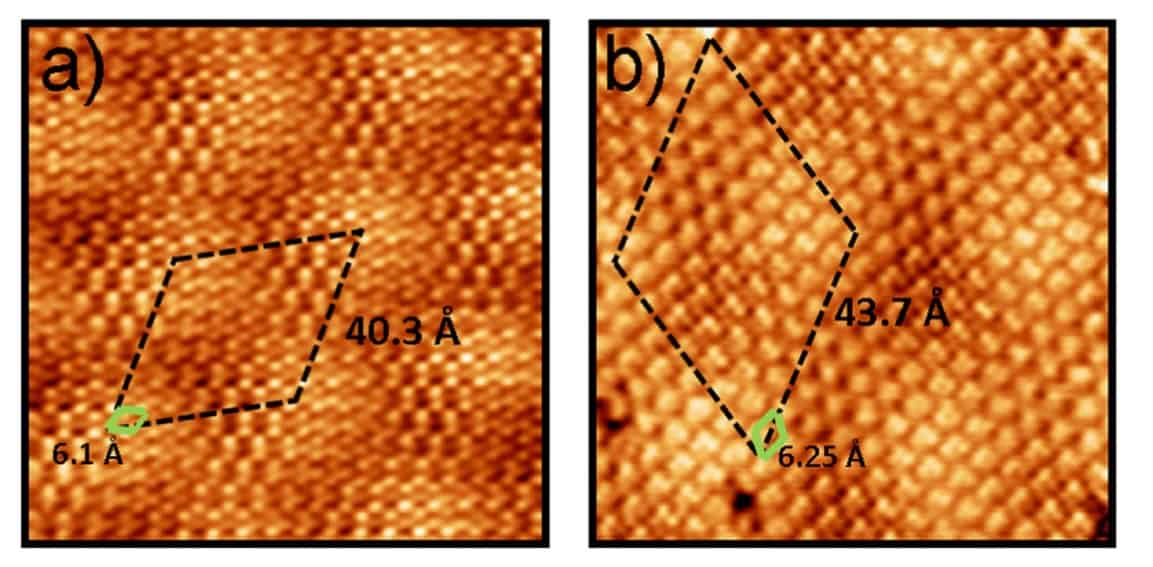
Perez and his team combined density functional simulations and scanning tunnelling microscopy observations to study the self-assembly of a molecular layer of triazine on graphene on different supporting materials. They found that although the intermolecular forces between the triazine were relatively strong compared with the interactions between the molecules and substrate, the self-assembly remained governed by a fine balance between the two.
In addition, despite using the most advanced density functional tools at their disposal, their calculations also revealed previously unknown fundamental limitations in the exchange-correlation functional, which led to discrepancies in the comparison of theory and experiment. “Spotting the limitations of these functionals, which are widely applied in many research fields, from catalysis to molecular electronics, is an important contribution of our work,” says Perez.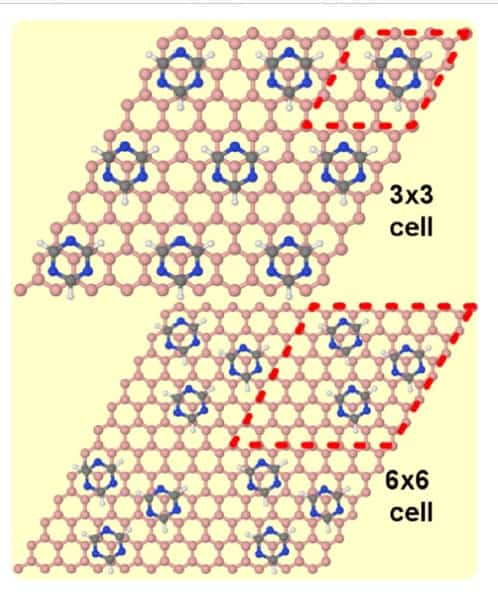 Ball-and-stick models for triazine on graphene. Courtesy of Nano Futures
Ball-and-stick models for triazine on graphene. Courtesy of Nano Futures
Ball-and-stick models for triazine on graphene. Courtesy of Nano Futures
The perfect molecule
Perez and colleagues focused their investigation on the self-assembly of triazine on account of numerous attributes that make it the “perfect candidate”. The molecule is like a benzene molecule – a ring of six carbons each bonded to one hydrogen atom on the outside of the ring and a carbon on either side – but in the case of triazine, three of the carbon atoms and associated hydrogens are replaced by nitrogen. This means that while remaining a small molecule, with a hexagonal structure to match graphene’s honeycomb lattice, the hydrogen bonds between the nitrogen atoms of one molecule and the hydrogen atoms of neighbouring molecules allows for strong intermolecular interactions, unlike benzene.
The researchers studied the self-assembly of triazine single molecule molecular layers on graphene, graphite and graphene on platinum, in particular the Pt(111) surface. “In spite of this strong intermolecular binding, the differences in interaction with the two substrates lead to markedly different self-assembled molecular layer periodicity,” says Perez.
In fact the researchers found that the molecule-substrate and the intermolecular interactions were of the same order. Interactions with the substrate influenced the orientation of the triazine, and intermolecular interactions then led to large moiré patterns – the fringes that result when overlaying two periodic patterns – as the balance of interactions wrestled with lattice mismatches not just with graphene but also the material beneath.Theory and experiment mismatch
It is the effect of the materials beneath the graphene that even state-of-the-art density functional calculations fail to produce. “So far, the community would tend to attribute this discrepancy with the experiments to the limitations of our description of the van der Waals interaction,” says Perez. “Our study, which includes the sophisticated MBD [many-body dispersion] approach, conclusively shows that this is not the case.”
Perez explains that short-range electronic correlations should be responsible for the effects of the material beneath the graphene. However none of the exchange-correlation functionals that the team considered seemed to describe them properly.  The authors
The authors
The authors
Peter Liljeroth, Group Leader of Atomic Scale Physics at Aalto University in Finland, is a world expert in this field, and not involved in the current research. He commented, “Density-functional theory calculations are extensively used in condensed-matter physics and materials science to predict new structures and materials and to understand existing experimental results in more detail. However, systems where the structure depends on a delicate balance between chemical and van der Waals interactions are difficult to capture quantitatively and experimental results on well-characterized systems are required as benchmarks for the theory. This extremely solid piece of work highlights the problems that still remain before DFT can reach true predictive power.”
Strengthened understanding of weakly interacting surfaces
With so much interest in maximizing the technological potential of graphene, you’d be forgiven for thinking the literature was rife with studies on any strategy that showed potential to manipulate the material’s fantastical properties. However, as Perez tells Physics World the self-assembly of molecular layers on graphene and other weakly interacting substrates remains little understood.
“From the experimental side, the preparation and characterization of SAMs [self-assembled molecular layers] on weakly interacting substrates is challenging,” explains Perez. He tells Physics World that most of the previous work in this line has concentrated on molecules adsorbed on noble metal substrates like gold, copper and silver. “In these cases, although the molecule-substrate interaction is small compared with other reactive metals, it is still strong enough to dominate the SAM formation.”
Next the Perez and his colleagues will be focusing their attention on understanding self-assembled molecular layers of single-stranded DNA and the role of water concentration in its structure. “This work is part of our research on the mechanical properties of proteins and nucleic acids in their native biological environment and is relevant for the development of biosensors to detect single mutations in DNA.”
Full details are reported in Nano Futures
8/9/2018 FROM PHYSICSWORLD.COM
Δεν υπάρχουν σχόλια:
Δημοσίευση σχολίου